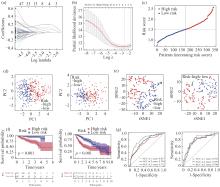
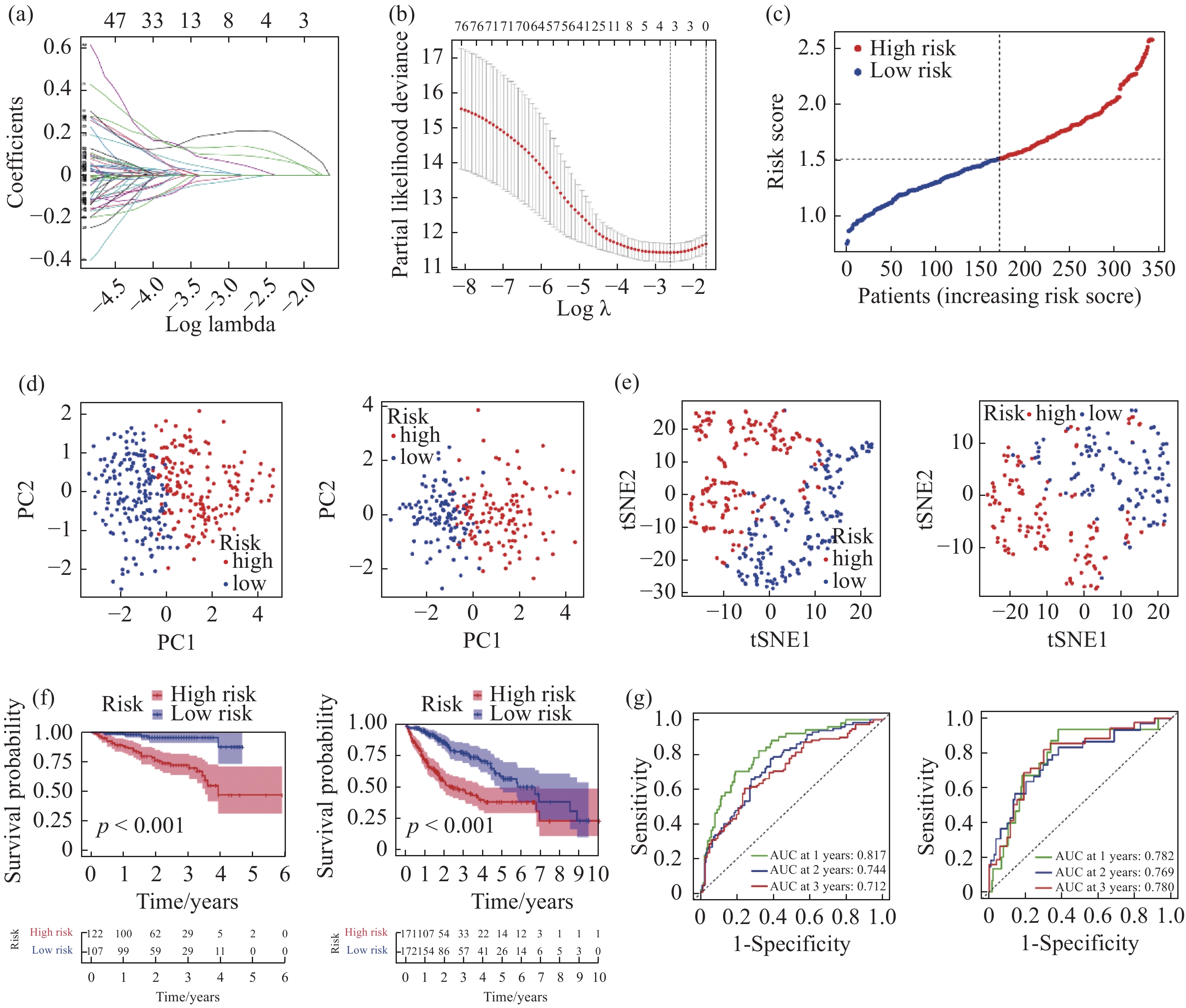
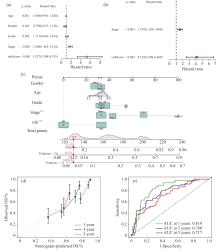
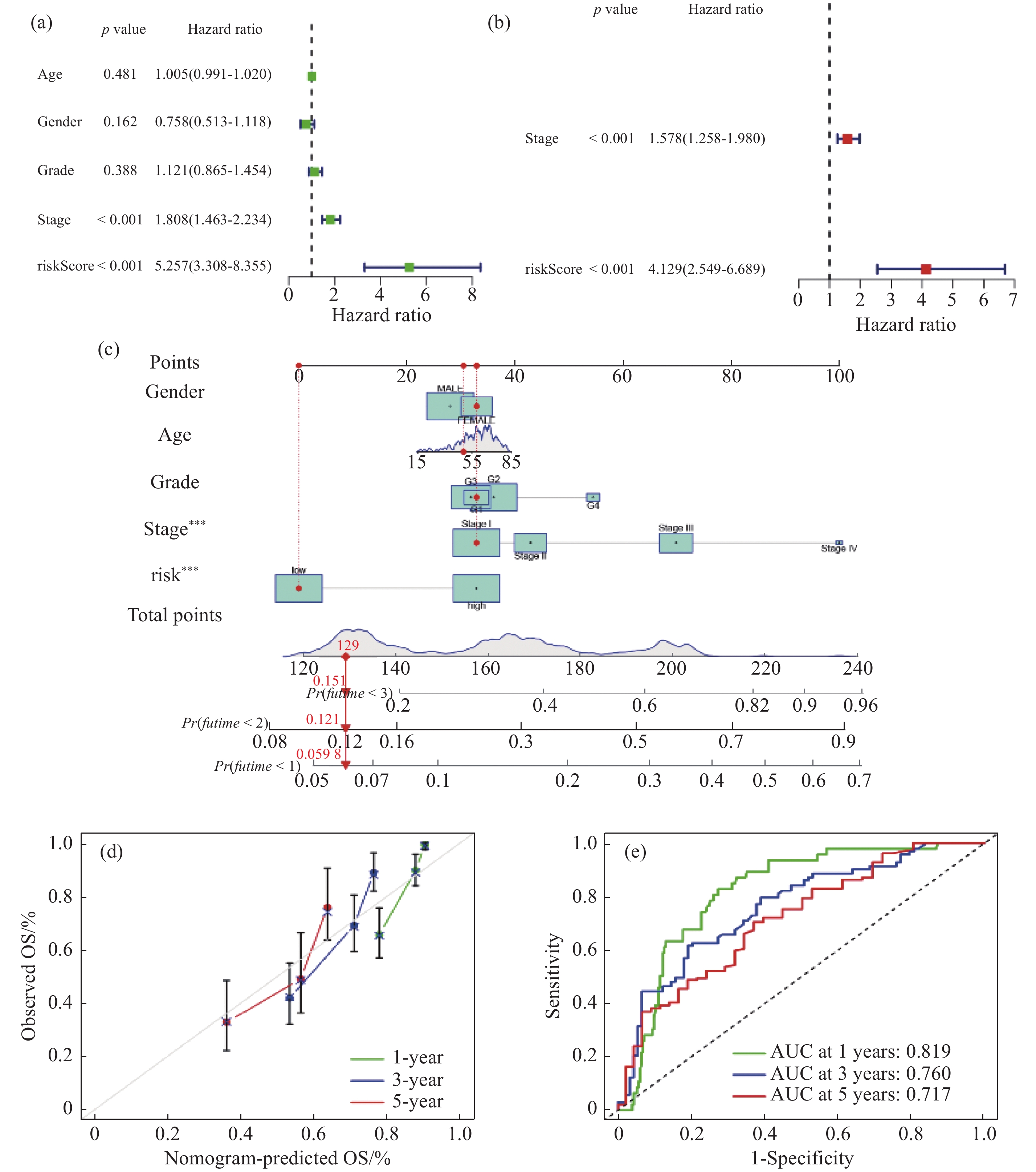
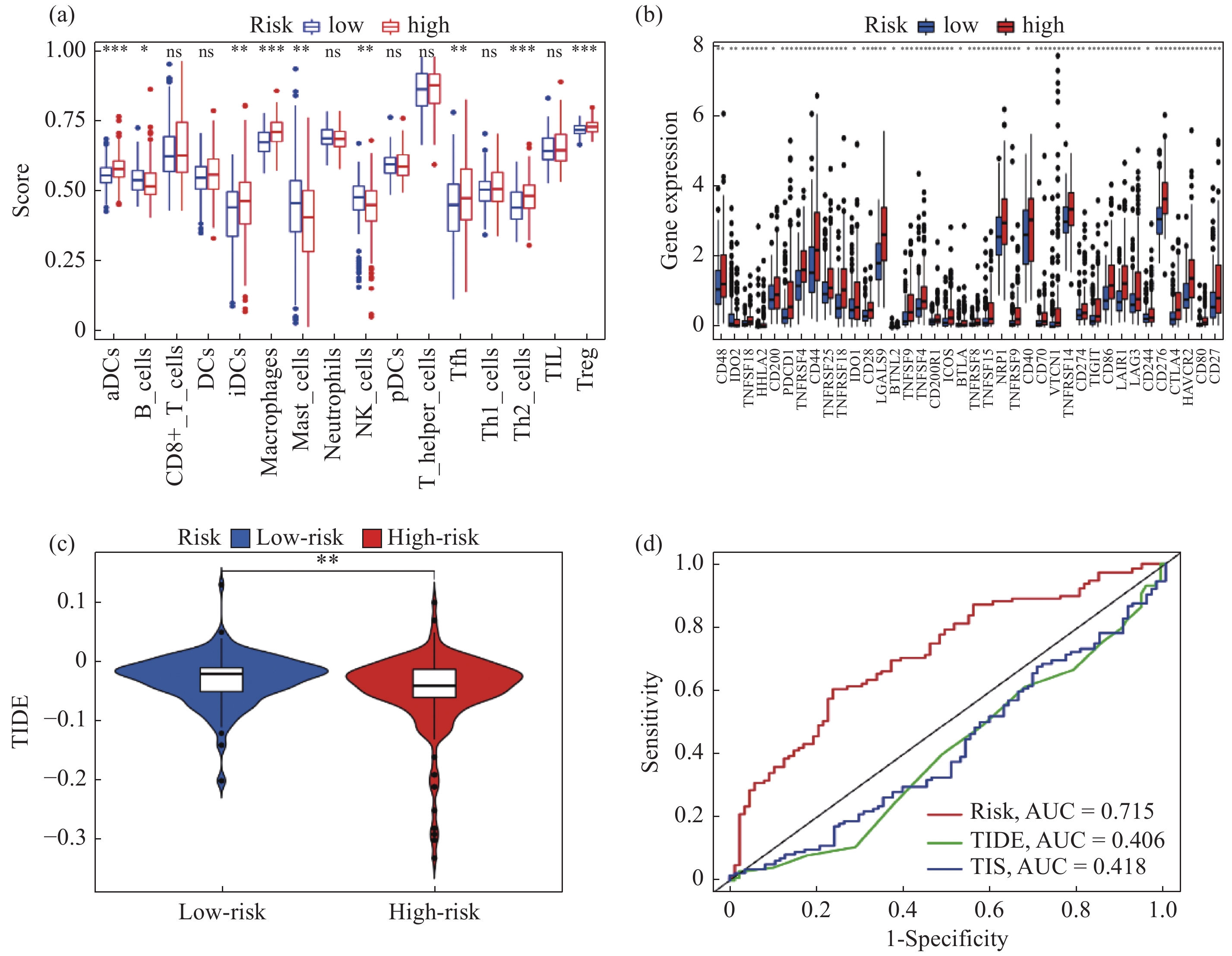
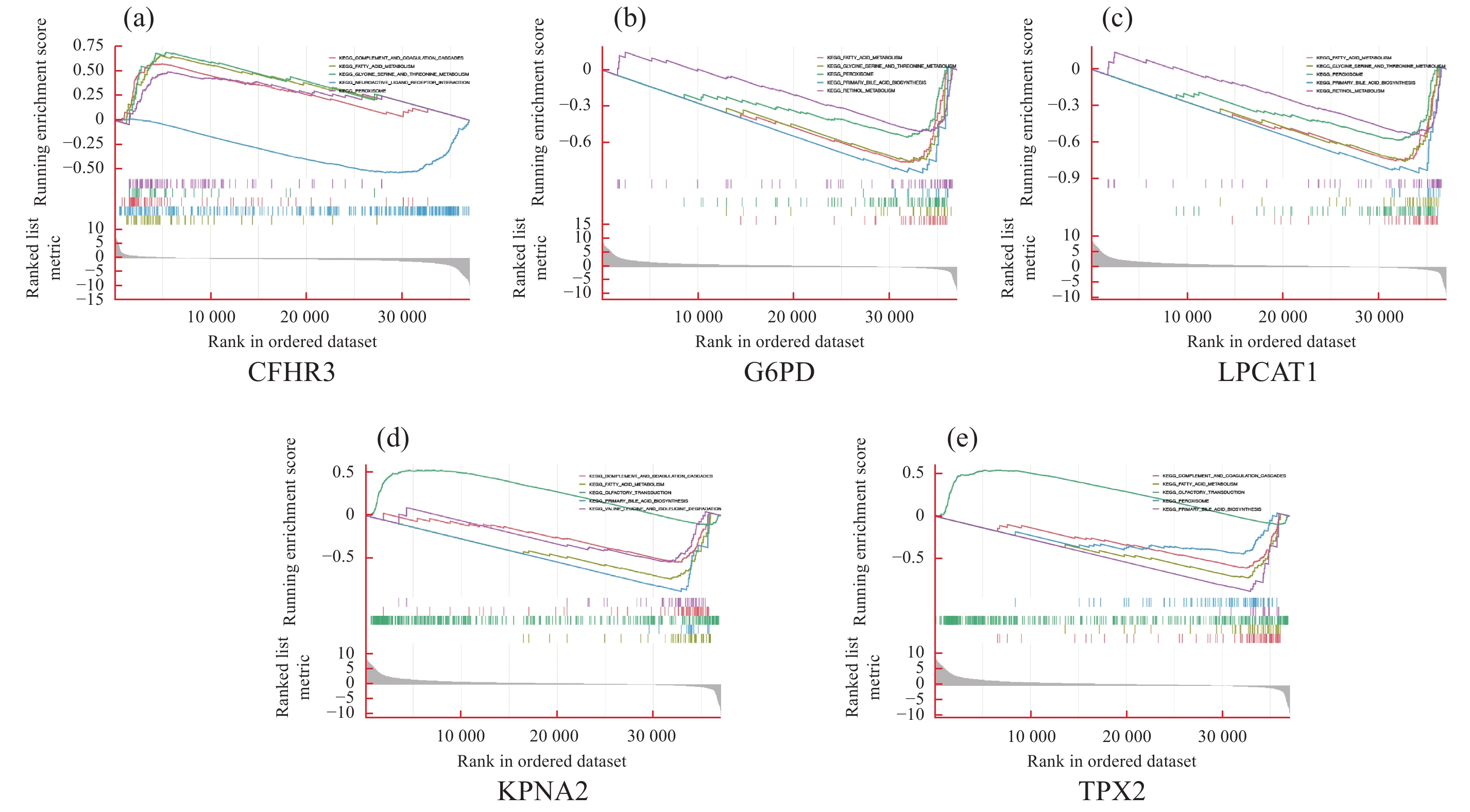
| 1 |
LLOVET J M, KELLEY R K, VILLANUEVA A, et al.. Hepatocellular carcinoma. Nature Reviews Disease Primers, 2021, 7 (1): 6.
|
| 2 |
SUNG H, FERLAY J, SIEGEL R L, et al.. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 2021, 71 (3): 209- 249.
|
| 3 |
中华人民共和国国家卫生健康委员会医政医管局. 原发性肝癌诊疗规范(2019年版) [J]. 中华肝脏病杂志, 2020, 28(2): 112-128.
|
| 4 |
ALLEMANI C, MATSUDA T, DI CARLO V, et al.. Global surveillance of trends in cancer survival 2000-14 (concord-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet, 2018, 391 (10125): 1023- 1075.
|
| 5 |
FUJIWARA N, FRIEDMAN S L, GOOSSENS N, et al.. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. Journal of Hepatology, 2018, 68 (3): 526- 549.
|
| 6 |
HEINRICH B, CZAUDERNA C, MARQUARDT J U.. Immunotherapy of hepatocellular carcinoma. Oncology Research & Treatment, 2018, 41 (5): 292- 297.
|
| 7 |
ZHANG B, TANG B, GAO J, et al.. A hypoxia-related signature for clinically predicting diagnosis, prognosis and immune microenvironment of hepatocellular carcinoma patients. Journal of Translational Medicine, 2020, 18 (1): 342.
|
| 8 |
LIU T, WU H, QI J, et al.. Seven immune-related genes prognostic power and correlation with tumor-infiltrating immune cells in hepatocellular carcinoma. Cancer Medicine, 2020, 9 (20): 7440- 7452.
|
| 9 |
CUI L, XUE H, WEN Z, et al.. Prognostic roles of metabolic reprogramming-associated genes in patients with hepatocellular carcinoma. Aging (Albany NY), 2020, 12 (21): 22199- 22219.
|
| 10 |
GUERRA A D, YEUNG O W H, QI X, et al.. The anti-tumor effects of m1 macrophage-loaded poly (ethylene glycol) and gelatin-based hydrogels on hepatocellular carcinoma. Theranostics, 2017, 7 (15): 3732- 3744.
|
| 11 |
ZHANG J, XI J, HUANG P, et al.. Comprehensive analysis identifies potential ferroptosis-associated mrna therapeutic targets in ovarian cancer. Frontiers in Medicine (Lausanne), 2021, 8, 644053.
|
| 12 |
HANZELMANN S, CASTELO R, GUINNEY J.. Gsva: Gene set variation analysis for microarray and rna-seq data. BMC Bioinformatics, 2013, 14, 7.
|
| 13 |
JIANG P, GU S, PAN D, et al.. Signatures of t cell dysfunction and exclusion predict cancer immunotherapy response. Nature Medicine, 2018, 24 (10): 1550- 1558.
|
| 14 |
OURA K, MORISHITA A, TANI J, et al. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: A review [J]. International Journal of Molecular Sciences, 2021, 22(11): 5801.
|
| 15 |
ZHANG Q, HE Y, LUO N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma [J]. Cell, 2019, 179(4): 829-845.
|
| 16 |
SANMAMED M F, CHEN L.. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell, 2018, 175 (2): 313- 326.
|
| 17 |
ZONGYI Y, XIAOWU L.. Immunotherapy for hepatocellular carcinoma. Cancer Letters, 2020, 470, 8- 17.
|
| 18 |
YANG H C, STERN A, CHIU D T.. G6PD: A hub for metabolic reprogramming and redox signaling in cancer. Biomedical Journal, 2021, 44 (3): 285- 292.
|
| 19 |
YANG H C, WU Y H, YEN W C, et al. The redox role of G6PD in cell growth, cell death, and cancer [J]. Cells, 2019, 8(9): 1055.
|
| 20 |
FANG Z, JIANG C, FENG Y, et al.. Effects of G6PD activity inhibition on the viability, ros generation and mechanical properties of cervical cancer cells. Biochim Biophys Acta, 2016, 1863 (9): 2245- 2254.
|
| 21 |
YANG C A, HUANG H Y, LIN C L, et al.. G6PD as a predictive marker for glioma risk, prognosis and chemosensitivity. Journal of Neuro-Oncology, 2018, 139 (3): 661- 670.
|
| 22 |
CHEN X, XU Z, ZHU Z, et al.. Modulation of G6PD affects bladder cancer via ros accumulation and the akt pathway in vitro. International Journal of Oncology, 2018, 53 (4): 1703- 1712.
|
| 23 |
ZHANG X, GAO F, AI H, et al.. TSP50 promotes hepatocyte proliferation and tumour formation by activating glucose-6-phosphate dehydrogenase (G6PD). Cell Proliferation, 2021, 54 (4): e13015.
|
| 24 |
POUW R B, GOMEZ DELGADO I, LOPEZ LERA A, et al.. High complement factor h-related (FHR)-3 levels are associated with the atypical hemolytic-uremic syndrome-risk allele CFHR3*B. Frontiers in Immunology, 2018, 9, 848.
|
| 25 |
SEOL H S, LEE S E, SONG J S, et al.. Complement proteins C7 and CFH control the stemness of liver cancer cells via LSF-1. Cancer Letters, 2016, 372 (1): 24- 35.
|
| 26 |
XIE J, KIRYLUK K, LI Y, et al.. Fine mapping implicates a deletion of CFHR1 and CFHR3 in protection from iga nephropathy in han chinese. Journal of the American Society of Nephrology, 2016, 27 (10): 3187- 3194.
|
| 27 |
LIU H, ZHANG L, WANG P.. Complement factor hrelated 3 overexpression affects hepatocellular carcinoma proliferation and apoptosis. Molecular Medicine Reports, 2019, 20 (3): 2694- 2702.
|
| 28 |
GRUPP K, SANADER S, SIRMA H, et al.. High lysophosphatidylcholine acyltransferase 1 expression independently predicts high risk for biochemical recurrence in prostate cancers. Molecular Oncology, 2013, 7 (6): 1001- 1011.
|
| 29 |
UEHARA T, KIKUCHI H, MIYAZAKI S, et al. Overexpression of lysophosphatidylcholine acyltransferase 1 and concomitant lipid alterations in gastric cancer [J]. Annals of Surgical Oncology, 2016, 23(Suppl 2): S206-S213.
|
| 30 |
JI W, PENG Z, SUN B, et al.. LPCAT1 promotes malignant transformation of hepatocellular carcinoma cells by directly suppressing STAT1. Annals of Surgical Oncology, 2021, 11, 678714.
|
| 31 |
ZHANG X, ZHANG J, GAO F, et al.. KPNA2-associated immune analyses highlight the dysregulation and prognostic effects of GRB2, nras, and their rna-binding proteins in hepatocellular carcinoma. Frontiers in Genetics, 2020, 11, 593273.
|
| 32 |
WARNER S L, STEPHENS B J, NWOKENKWO S, et al.. Validation of TPX2 as a potential therapeutic target in pancreatic cancer cells. Clinical Cancer Research, 2009, 15 (21): 6519- 6528.
|
| 33 |
HSU P K, CHEN H Y, YEH Y C, et al.. TPX2 expression is associated with cell proliferation and patient outcome in esophageal squamous cell carcinoma. Journal of Gastroenterology, 2014, 49 (8): 1231- 1240.
|
| 34 |
HSU C W, CHEN Y C, SU H H, et al.. Targeting TPX2 suppresses the tumorigenesis of hepatocellular carcinoma cells resulting in arrested mitotic phase progression and increased genomic instability. Journal of Cancer, 2017, 8 (8): 1378- 1394.
|
| 35 |
WANG Y, WANG H, YAN Z, et al.. The critical role of dysregulated Hh-FOXM1-TPX2 signaling in human hepatocellular carcinoma cell proliferation. Cell Communication and Signaling, 2020, 18 (1): 116.
|
| 36 |
KIM K, PARK S, PARK S Y, et al.. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Medicine, 2020, 12 (1): 22.
|
| 37 |
JIANG R, TANG J, CHEN Y, et al.. The long noncoding rna lnc-EGFR stimulates t-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nature Communications, 2017, (8): 15129.
|
| 38 |
DONISI C, PUZZONI M, ZIRANU P, et al.. Immune checkpoint inhibitors in the treatment of hcc. Frontiers in Oncology, 2020, 10, 601240.
|
 )
)
 中文核心期刊
中文核心期刊