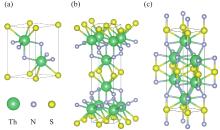
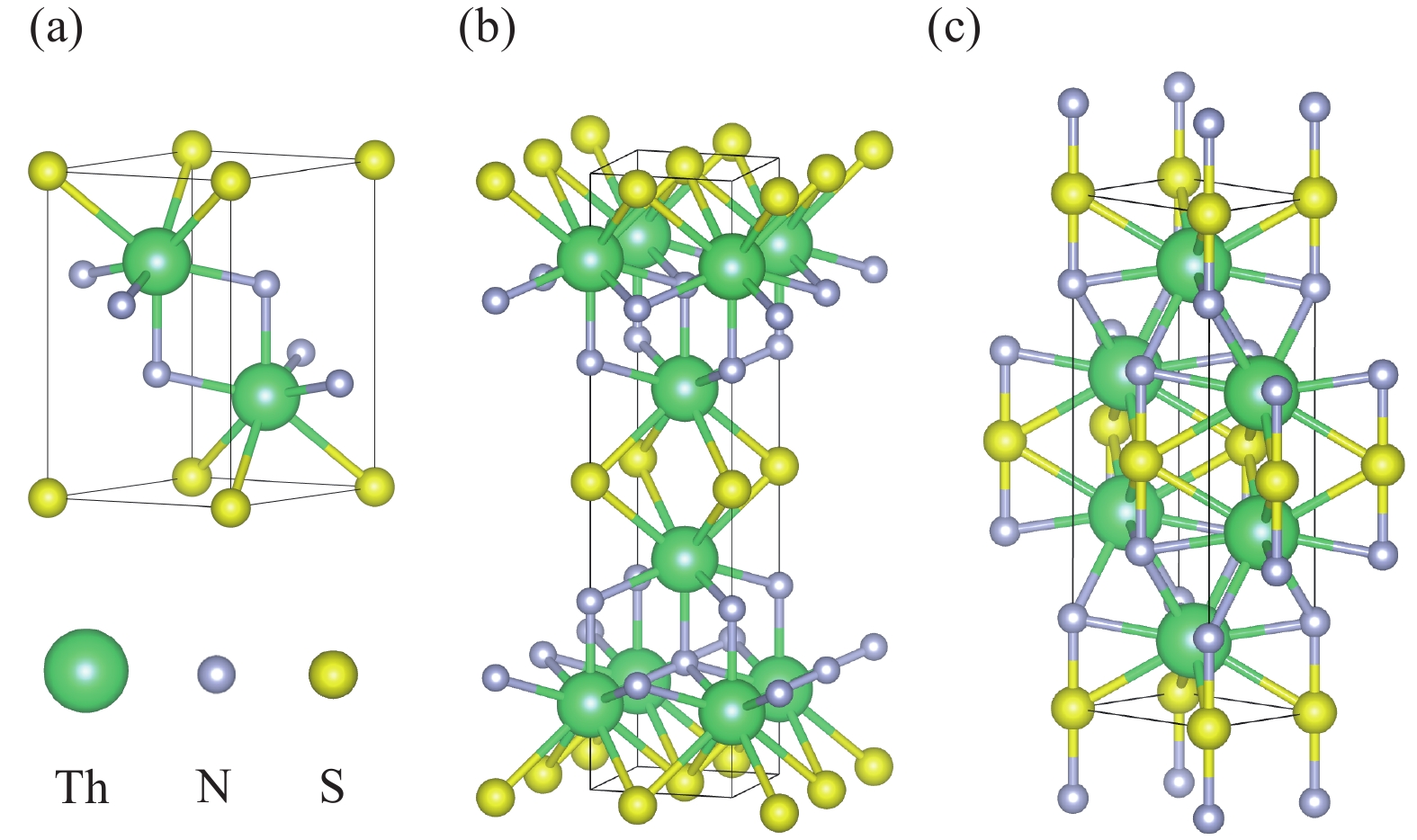
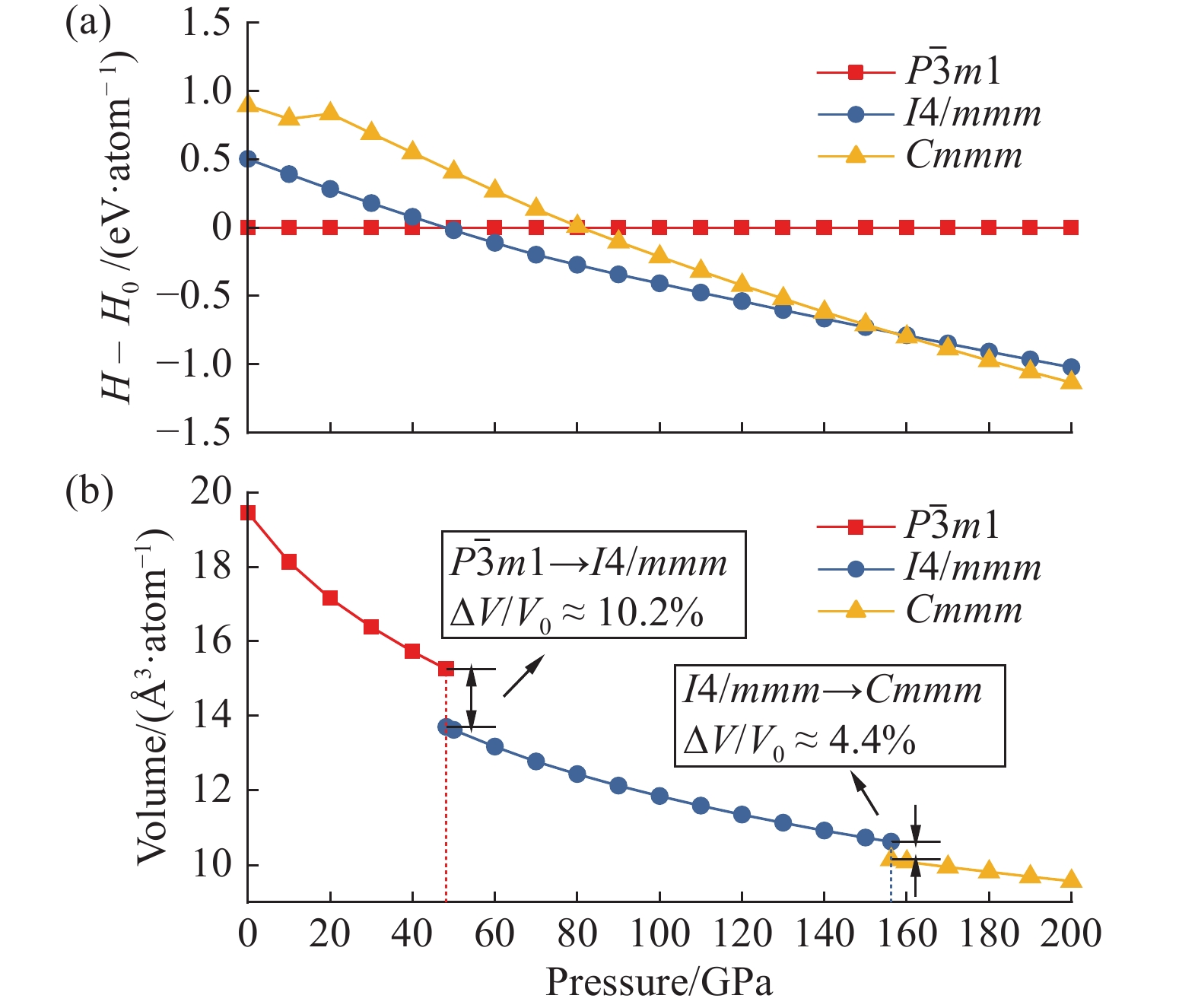
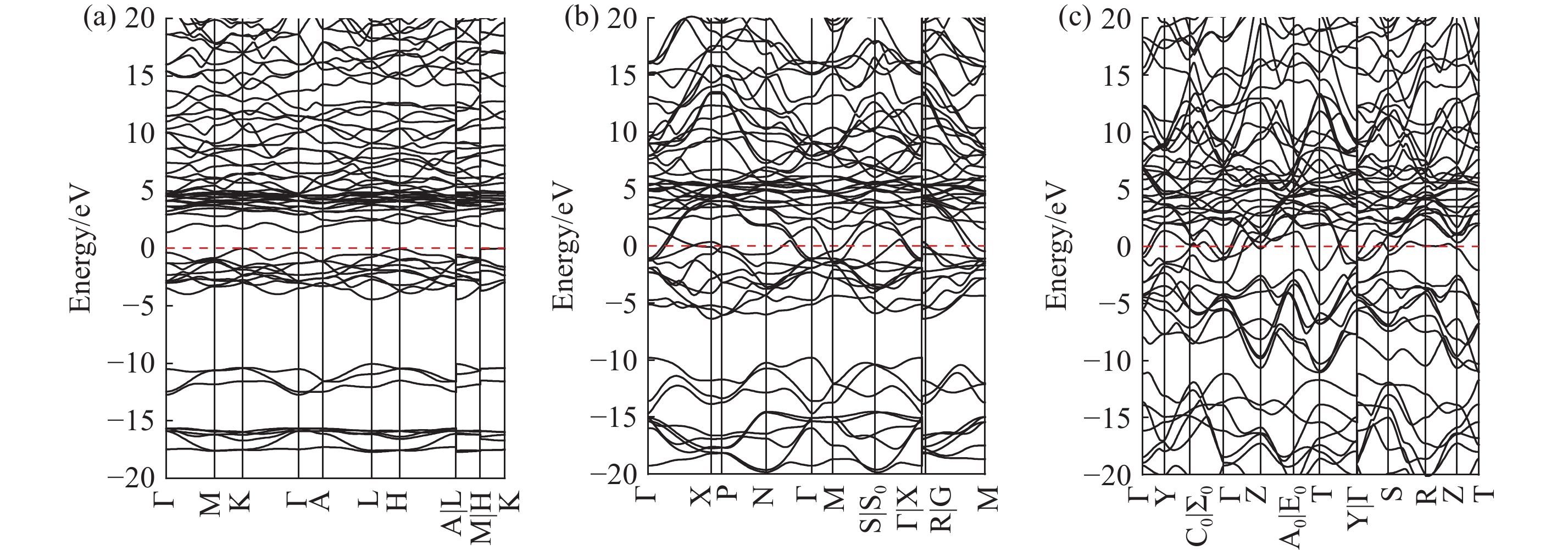
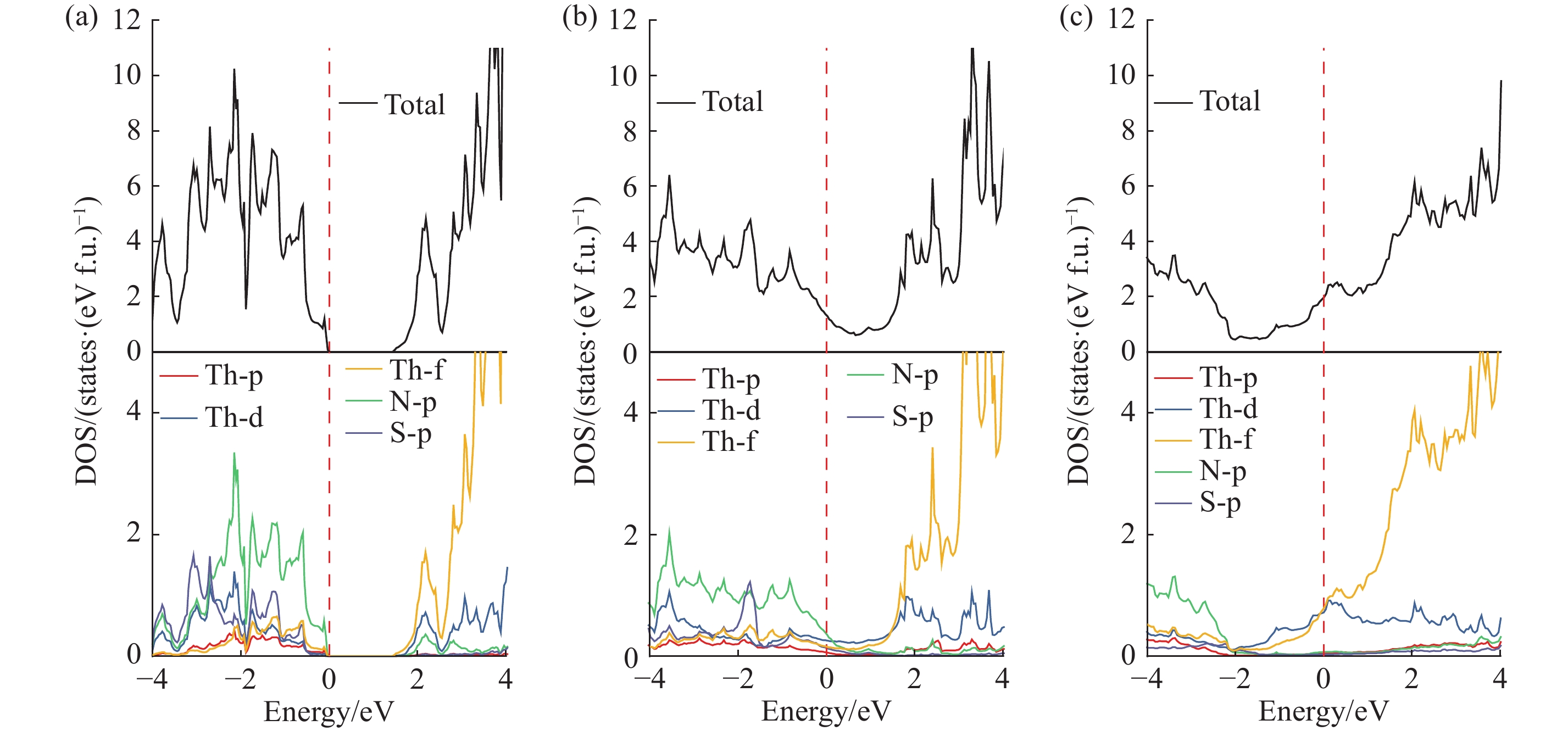
| 1 |
ANDERSEN M B, ELLIOTT T, FREYMUTH H, et al.. The terrestrial uranium isotope cycle. Nature, 2015, 517 (7534): 356- 359.
|
| 2 |
BAGLA P.. Thorium seen as nuclear’s new frontier. Science, 2015, 350 (6262): 726- 727.
|
| 3 |
GRIMES R W, NUTTALL W J.. Generating the option of a two-stage nuclear renaissance. Science, 2010, 329 (5993): 799- 803.
|
| 4 |
GUO Y L, QIU W J, KE X Z, et al.. A new phase of ThC at high pressure predicted from a first-principles study. Physics Letters A, 2015, 379 (26/27): 1607- 1611.
|
| 5 |
YU C, LIN J, HUAI P, et al.. Structural phase transition of ThC under high pressure. Scientific Reports, 2017, (7): 96.
|
| 6 |
GUO Y L, YU C, LIN J, et al.. Pressure-induced structural transformations and polymerization in ThC2. Scientific Reports, 2017, (7): 45872.
|
| 7 |
GERWARD L, OLSEN J S, BENEDICT U, et al.. The crystal structure and the equation of state of thorium nitride for pressures up to 47 GPa. Journal of Applied Crystallography, 1985, 18 (5): 339- 341.
|
| 8 |
MODAK P, VERMA A K.. First-principles investigation of electronic, vibrational, elastic, and structural properties of ThN and UN up to 100 GPa. Physical Review B, 2011, 84 (2): 024108.
|
| 9 |
IDIRI M, LE BIHAN T, HEATHMAN S, et al.. Behavior of actinide dioxides under pressure: UO2 and ThO2. Physical Review B, 2004, 70 (1): 014113.
|
| 10 |
LIANG B Y, ANDREWS L.. Matrix infrared spectra and quasirelativistic DFT studies of ThS and ThS2. The Journal of Physical Chemistry A, 2002, 106 (16): 4038- 4041.
|
| 11 |
GUO Y L, WANG C Y, QIU W J, et al.. Structural and electronic phase transitions of ThS2 from first-principles calculations. Physical Review B, 2016, 94 (13): 134104.
|
| 12 |
ARIF KHALIL R M, HUSSAIN M I, SAEED N, et al.. The prediction of structural, electronic, optical and vibrational behavior of ThS2 for nuclear fuel applications: A DFT study. Optical and Quantum Electronics, 2021, 53, 11.
|
| 13 |
SAHOO B D, JOSHI K D, KAUSHIK T C.. High pressure structural stability of ThN: Ab-initio study. Journal of Nuclear Materials, 2019, 521, 161- 166.
|
| 14 |
SAHOO B D, JOSHI K D, KAUSHIK T C.. Structural, elastic, vibrational, thermophysical properties and pressure-induced phase transitions of ThN2, Th2N3, and Th3N4: An ab initio investigation. Journal of Applied Physics, 2020, 128 (3): 035902.
|
| 15 |
ZHANG Y, GUO Y L, LIAO Z G, et al.. Ab initio investigation of pressure-induced structural transitions and electronic evolution of Th3N4. High Pressure Research, 2020, 40 (2): 267- 282.
|
| 16 |
BENZ R, ZACHARIASEN W H.. Crystal structure of the compounds U2N2X and Th2(N, O)2 with X= P, S, As and Se. Acta Crystallographica Section B: Structural Crystallography and Crystal Chemistry, 1969, 25 (2): 294- 296.
|
| 17 |
WANG Y C, LV J, ZHU L, et al.. CALYPSO: A method for crystal structure prediction. Computer Physics Communications, 2012, 183 (10): 2063- 2070.
|
| 18 |
MADDOX J.. Crystals from first principles. Nature, 1988, 335, 201- 201.
|
| 19 |
LONIE D C, ZUREK E.. XTALOPT version r7: An open-source evolutionary algorithm for crystal structure prediction. Computer Physics Communications, 2011, 182 (10): 2305- 2306.
|
| 20 |
WANG Y C, LV J, ZHU L, et al.. Crystal structure prediction via particle-swarm optimization. Physical Review B, 2010, 82 (9): 094116.
|
| 21 |
KRESSE G, FURTHMÜLLER J.. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 1996, 54 (16): 11169.
|
| 22 |
KRESSE G, FURTHMÜLLER J.. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science, 1996, 6 (1): 15- 50.
|
| 23 |
PERDEW J P, BURKE K, ERNZERHOF M.. Generalized gradient approximation made simple. Physical Review Letters, 1996, 77 (18): 3865- 3868.
|
| 24 |
TOGO A, OBA F, TANAKA I.. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Physical Review B, 2008, 78 (13): 134106.
|
| 25 |
PARLINSKI K, LI Z Q, KAWAZOE Y.. First-principles determination of the soft mode in cubic ZrO2. Physical Review Letters, 1997, 78 (21): 4063- 4066.
|
| 26 |
MATAR S F, KFOURY C N.. Combined crystal chemistry and DFT studies of ThNCl and Th2N2X (X: chalcogen) behaving as pseudo-binaries. Solid State Sciences, 2018, 76, 1- 7.
|
| 27 |
COCHRAN W.. Crystal stability and the theory of ferroelectricity. Advances in Physics, 1960, 9 (36): 387- 423.
|
| 28 |
LE PAGE Y, SAXE P.. Symmetry-general least-squares extraction of elastic data for strained materials from ab initio calculations of stress. Physical Review B, 2002, 65 (10): 104104.
|
| 29 |
MOUHAT F, COUDERT F X.. Necessary and sufficient elastic stability conditions in various crystal systems. Physical Review B, 2014, 90 (22): 224104.
|
| 30 |
WATT J P, PESELNICK L.. Clarification of the Hashin‐Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. Journal of Applied Physics, 1980, 51 (3): 1525- 1531.
|
| 31 |
CHUNG D H, BUESSEM W R.. The elastic anisotropy of crystals. Journal of Applied Physics, 1967, 38 (5): 2010- 2012.
|
| 32 |
HILL R.. The elastic behaviour of a crystalline aggregate. Proceedings of the Physical Society, Section A, 1952, 65 (5): 349- 354.
|
| 33 |
PUGH S F.. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 1954, 45 (367): 823- 843.
|
| 34 |
RANGANATHAN S I, OSTOJA-STARZEWSKI M.. Universal elastic anisotropy index. Physical Review Letters, 2008, 101 (5): 055504.
|
| 35 |
KOSCIELSKI L A, RINGE E, VAN DUYNE R P, et al.. Single-crystal structures, optical absorptions, and electronic distributions of thorium oxychalcogenides ThOQ (Q= S, Se, Te). Inorganic Chemistry, 2012, 51 (15): 8112- 8118.
|
| 36 |
SHEIN I R, SHEIN K I, IVANOVSKII A L.. First-principle study of B1-like thorium carbide, nitride and oxide. Journal of Nuclear Materials, 2006, 353 (1/2): 19- 26.
|
| 37 |
DAROCA D P, JAROSZEWICZ S, LLOIS A M, et al.. Phonon spectrum, mechanical and thermophysical properties of thorium carbide. Journal of Nuclear Materials, 2013, 437 (1/2/3): 135- 138.
|
 )
)
 中文核心期刊
中文核心期刊