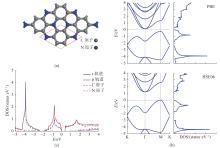
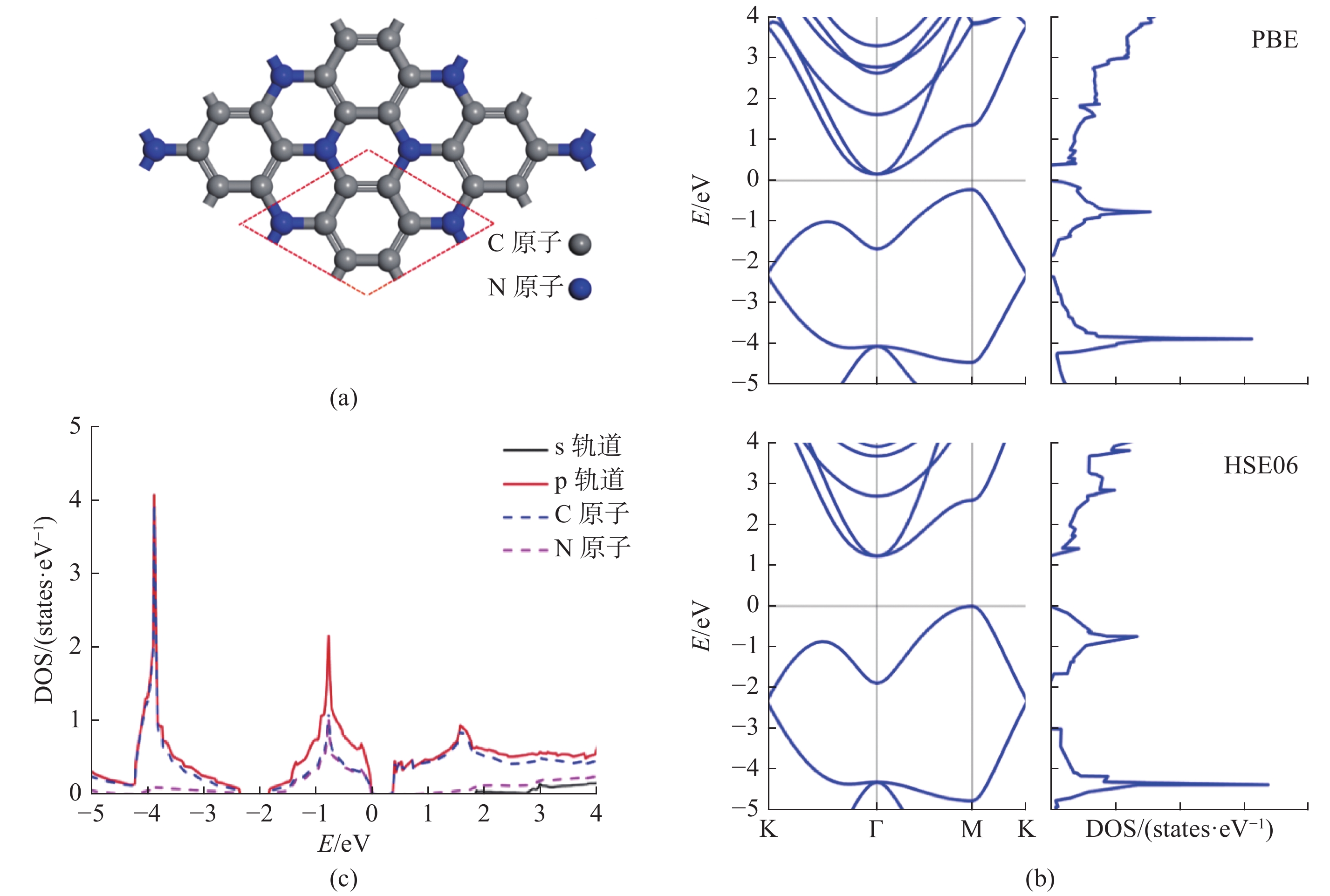
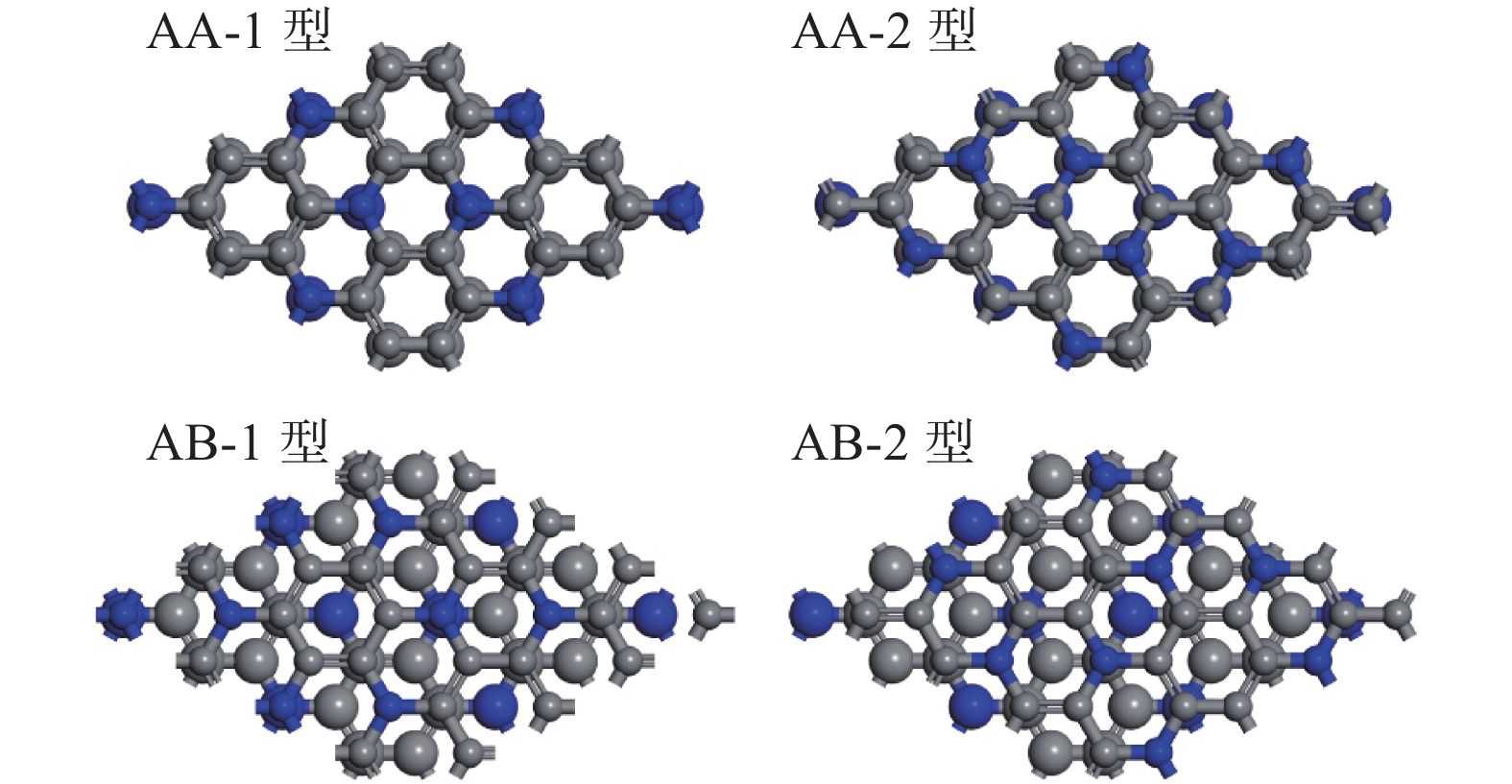
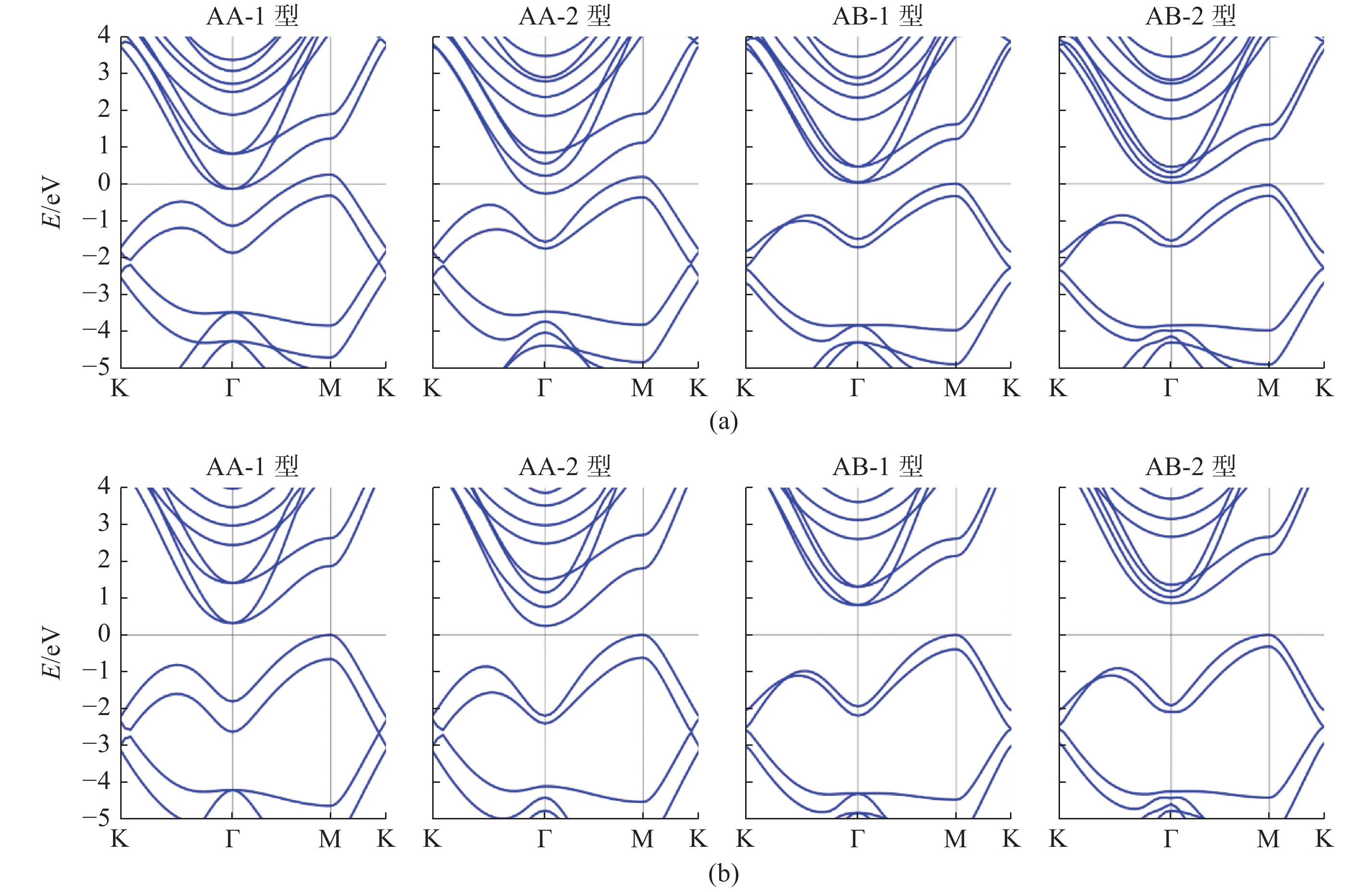
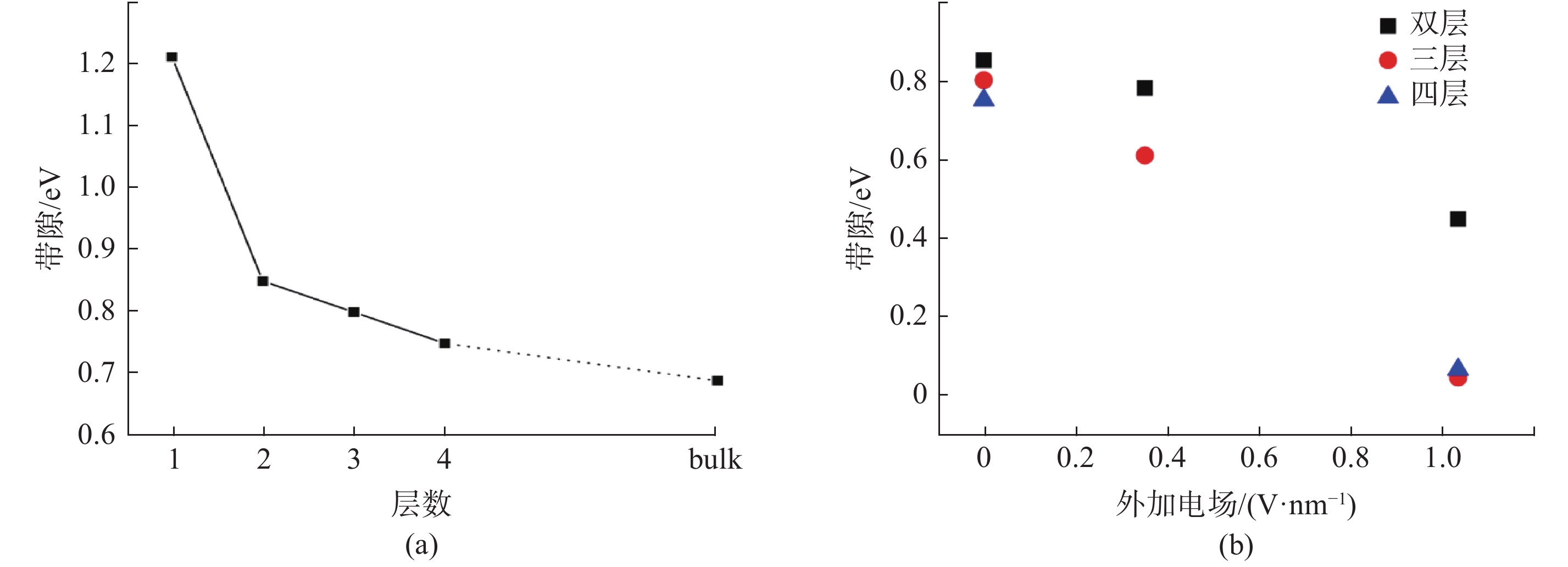
| 1 |
NOVOSELOV K S, GEIM A K, MOROZOV S V, et al. Electric field effect in atomically thin carbon films. Science, 2004, 306 (5696): 666- 669.
|
| 2 |
GEIM A K, NOVOSELOV K S. The rise of graphene. Nature Materials, 2007, 6 (3): 183- 191.
|
| 3 |
KIM K, CHOI J Y, KIM T, et al. A role for graphene in silicon-based semiconductor devices. Nature, 2011, 479 (7373): 338- 344.
|
| 4 |
SCHWIERZ F. Graphene transistors. Nature Nanotechnology, 2010, 5 (7): 487- 496.
|
| 5 |
MERIC I, HAN M Y, YOUNG A F, et al. Current saturation in zero-bandgap, top-gated graphene field-effect transistors. Nature Nanotechnology, 2008, 3 (11): 654- 659.
|
| 6 |
WANG X C, MAEDA K, THOMAS A, et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nature Materials, 2009, 8 (1): 76- 80.
|
| 7 |
MAHMOOD J, LEE E K, JUNG M, et al. Nitrogenated holey two-dimensional structures. Nature Communications, 2015, (6): 6486.
|
| 8 |
YANG S W, LI W, YE C C, et al. C3N—A 2D crystalline, hole-free, tunable-narrow-bandgap semiconductor with ferromagnetic properties . Advanced Materials, 2017, 29 (16): 1605625.
|
| 9 |
KRESSE G, FURTHMÜLLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 1996, 54 (16): 11169- 11186.
|
| 10 |
PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple. Physical Review Letters, 1996, 77 (18): 3865- 3868.
|
| 11 |
HEYD J, SCUSERIA G E, ERNZERHOF M. Hybrid functionals based on a screened Coulomb potential. Journal of Chemical Physics, 2003, 118 (18): 8207- 8215.
|
| 12 |
BLÖCHL P E. Projector augmented-wave method. Physical Review B, 1994, 50 (24): 17953- 17979.
|
| 13 |
MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations. Physical Review B, 1976, 13 (12): 5188- 5192.
|
| 14 |
HU Q K, WU Q H, WANG H Y, et al. First-principles studies of structural and electronic properties of layered C3N phases . Physica Status Solidi B, 2012, 249 (4): 784- 788.
|
| 15 |
ZHOU X D, FENG W X, GUAN S, et al. Computational characterization of monolayer C3N: A two-dimensional nitrogen-graphene crystal . Journal of Materials Research, 2017, 32 (15): 2993- 3001.
|
| 16 |
WATANABE K, TANIGUCHI T, KANDA H. Ultraviolet luminescence spectra of boron nitride single crystals grown under high pressure and high temperature. Physica Status Solidi A, 2004, 201 (11): 2561- 2565.
|
| 17 |
ELLIS J K, LUCERO M J, SCUSERIA G E. The indirect to direct band gap transition in multilayered MoS2 as predicted by screened hybrid density functional theory . Applied Physics Letters, 2011, 99 (26): 268901.
|
| 18 |
FERRARI A C, MEYER J C, SCARDACI V, et al. Raman spectrum of graphene and graphene layers. Physical Review Letters, 2006, 97 (18): 187401.
|
| 19 |
MORTAZAVI B. Ultra high stiffness and thermal conductivity of graphene like C3N . Carbon, 2017, 118, 25- 34.
|
| 20 |
BALOG R, JØRGENSEN B, NILSSON L, et al. Bandgap opening in graphene induced by patterned hydrogen adsorption. Nature Materials, 2010, 9 (4): 315- 319.
|
 )
)
 中国综合性科技类核心期刊(北大核心)
中国综合性科技类核心期刊(北大核心)