 中文核心期刊
中文核心期刊J* E* C* N* U* N* S* ›› 2025, Vol. 2025 ›› Issue (3): 100-108.doi: 10.3969/j.issn.1000-5641.2025.03.012
• Physics and Electronics • Previous Articles
Yue XIANG1, Min LIANG1, Yisu WANG1, Wenhui XIE1,2,*( )
)
Received:2024-11-14
Online:2025-05-25
Published:2025-05-28
Contact:
Wenhui XIE
E-mail:whxie@phy.ecnu.edu.cn
CLC Number:
Yue XIANG, Min LIANG, Yisu WANG, Wenhui XIE. Computational study of rutile-structured RuO2 using NMTO method[J]. J* E* C* N* U* N* S*, 2025, 2025(3): 100-108.
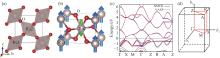
Fig.1
(a)Crystal structure and (b)Antiferromagnetic ordering diagram of rutile-structured RuO2, (c)Comparison of the band structure obtained by NMTO (blue) and VASP (red), (d)Selection path of high-symmetry K-points"
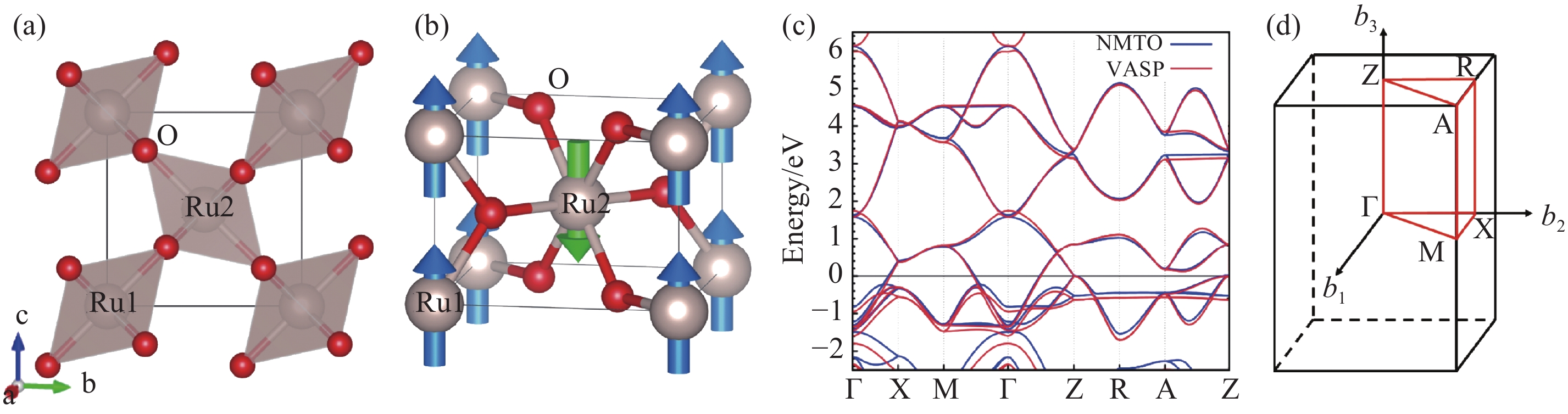
Fig.2
Fermi surface of RuO2 obtained via GGA approximation (left: NMTO, right: VASP)"
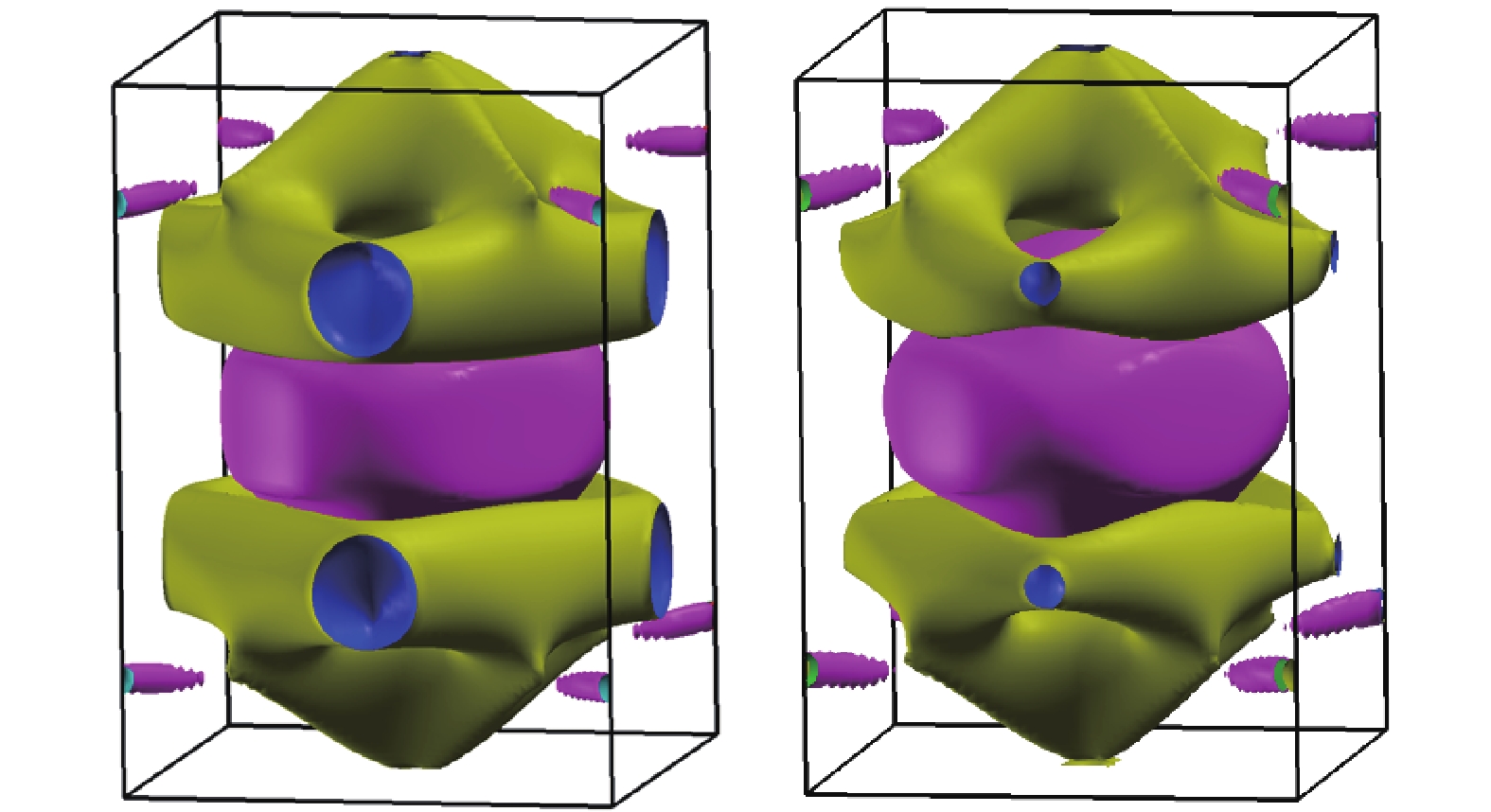

Fig.3
GGA + U calculation results for RuO2"
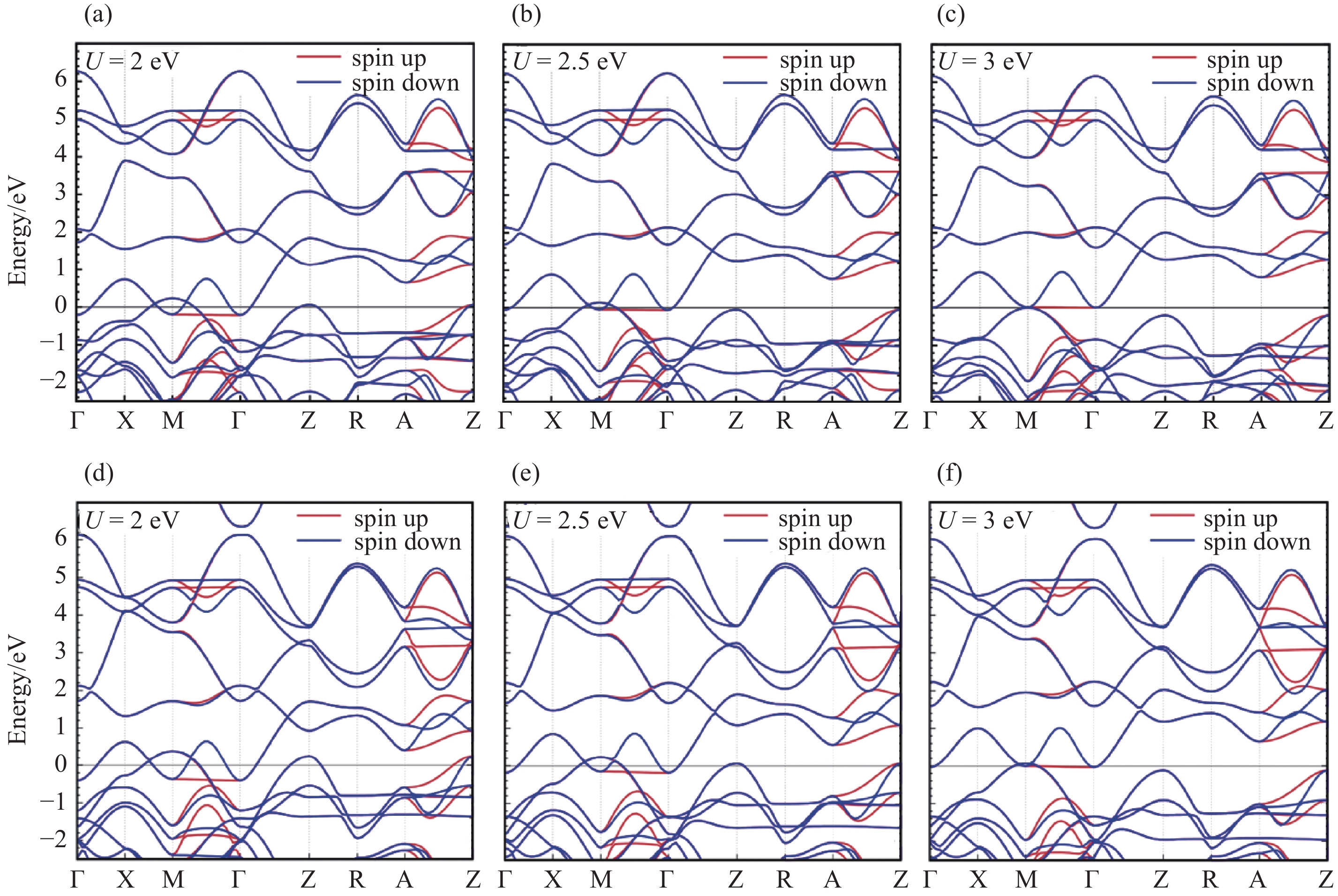
Fig.4
Spatial distribution of the five d orbitals of Ru calculated via NMTO"
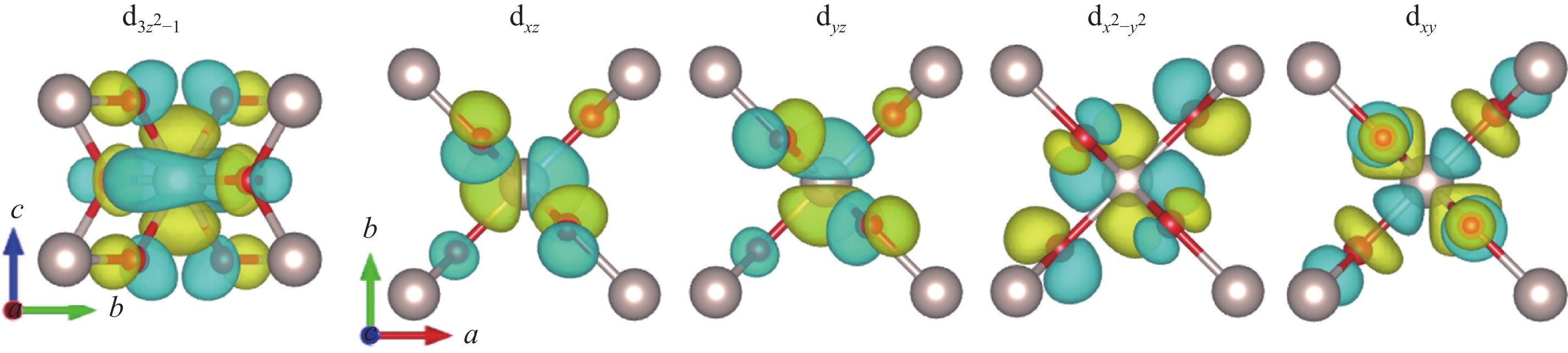

Fig.5
RuO2 band structure near the Fermi surface fitted using the NMTO downfolding method(a), and using Wannier90(b)"

| 1 | OVER H.. Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: From fundamental to applied research. Chemical Reviews, 2012, 112 (6): 3356- 3426. |
| 2 | UCHIDA M, NOMOTO T, MUSASHI M, et al.. Superconductivity in uniquely strained RuO2 films. Physical Review Letters, 2020, 125 (14): 147001. |
| 3 | RUF J P, PAIK H, SCHREIBER N J, et al.. Strain-stabilized superconductivity. Nature Communications, 2021, 12 (1): 59. |
| 4 | RYDEN W D, LAWSON A W.. Magnetic susceptibility of IrO2 and RuO2. The Journal of Chemical Physics, 1970, 52 (12): 6058- 6061. |
| 5 | FENG Z X, ZHOU X R, ŠMEJKAL L, et al.. An anomalous Hall effect in altermagnetic ruthenium dioxide. Nature Electronics, 2022, 5 (11): 735- 743. |
| 6 | BERLIJN T, SNIJDERS P C, DELAIRE O, et al.. Itinerant antiferromagnetism in RuO2. Physical Review Letters, 2017, 118 (7): 077201. |
| 7 | LOVESEY S W, KHALYAVIN D D, VAN DER LAAN G.. Magnetic properties of RuO2 and charge-magnetic interference in Bragg diffraction of circularly polarized x-rays. Physical Review B, 2022, 105 (1): 014403. |
| 8 | GONZÁLEZ-HERNÁNDEZ R, ŠMEJKAL L, VÝBORNÝ K, et al.. Efficient electrical spin splitter based on nonrelativistic collinear antiferromagnetism. Physical Review Letters, 2021, 126 (12): 127701. |
| 9 | CHI B, JIANG L, ZHU Y, et al.. Crystal facet orientated altermagnets for detecting ferromagnetic and antiferromagnetic states by giant tunneling magnetoresistance effect. Physical Review Applied, 2024, 21 (3): 034038. |
| 10 | SUN Y, ZHANG Y, LIU C X, et al.. Dirac nodal lines and induced spin Hall effect in metallic rutile oxides. Physical Review B, 2017, 95 (23): 235104. |
| 11 | JOVIC V, KOCH R J, PANDA S K, et al.. Dirac nodal lines and flat-band surface state in the functional oxide RuO2. Physical Review B, 2018, 98 (24): 241101. |
| 12 | ZHOU X, FENG W, YANG X, et al.. Crystal chirality magneto-optical effects in collinear antiferromagnets. Physical Review B, 2021, 104 (2): 024401. |
| 13 | LIU J, ZHAN J, LI T, et al.. Absence of altermagnetic spin splitting character in rutile oxide RuO2. Physical Review Letters, 2024, 133 (17): 176401. |
| 14 | ŠMEJKAL L, GONZÁLEZ-HERNÁNDEZ R, JUNGWIRTH T, et al.. Crystal time-reversal symmetry breaking and spontaneous Hall effect in collinear antiferromagnets. Science Advances, 2020, 6 (23): eaaz8809. |
| 15 | ŠMEJKAL L, HELLENES A B, GONZÁLEZ-HERNÁNDEZ R, et al.. Giant and tunneling magnetoresistance in unconventional collinear antiferromagnets with nonrelativistic spin-momentum coupling. Physical Review X, 2022, 12 (1): 011028. |
| 16 | BAI H, HAN L, FENG X Y, et al.. Observation of spin splitting torque in a collinear antiferromagnet RuO2. Physical Review Letters, 2022, 128 (19): 197202. |
| 17 | BOSE A, SCHREIBER N J, JAIN R, et al.. Tilted spin current generated by the collinear antiferromagnet ruthenium dioxide. Nature Electronics, 2022, 5 (5): 267- 274. |
| 18 | KARUBE S, TANAKA T, SUGAWARA D, et al.. Observation of spin-splitter torque in collinear antiferromagnetic RuO2. Physical Review Letters, 2022, 129 (13): 137201. |
| 19 | SHAO D F, ZHANG S H, XIAO R C, et al.. Spin-neutral tunneling anomalous Hall effect. Physical Review B, 2022, 106 (18): L180404. |
| 20 | WADLEY P, HOWELLS B, ŽELEZNÝ J, et al.. Electrical switching of an antiferromagnet. Science, 2016, 351 (6273): 587- 590. |
| 21 | BALTZ V, MANCHON A, TSOI M, et al.. Antiferromagnetic spintronics. Reviews of Modern Physics, 2018, 90 (1): 015005. |
| 22 | MAURYA V, SHARMA G, JOSHI K B.. First-principles characterisation of structural and electronic properties of some RuO2 crystals. Physica Scripta, 2021, 96 (5): 055807. |
| 23 | TANAKA K, NOMOTO T, ARITA R.. First-principles study of the tunnel magnetoresistance effect with Cr-doped RuO2 electrode. Physical Review B, 2024, 110 (6): 064433. |
| 24 | NOHARA Y, ANDERSEN O K.. Interpolation across a muffin-tin interstitial using localized linear combinations of spherical waves. Physical Review B, 2016, 94 (8): 085148. |
| 25 | YAMASAKI A, CHIONCEL L, LICHTENSTEIN A I, et al.. Model Hamiltonian parameters for half-metallic ferromagnets NiMnSb and CrO2. Physical Review B, 2006, 74 (2): 024419. |
| 26 | SAHA-DASGUPTA T, ANDERSEN O K, NUSS J, et al. Electronic structure of V2O3: Wannier orbitals from LDA-NMTO calculations[EB/OL]. (2009-07-16)[2024-09-27]. https://arxiv.org/pdf/0907.2841. |
| 27 | ANDERSEN O K. NMTOs and their Wannier functions [C]// Correlated Electrons: From Model to Materials. Jülich: Forschungszentrum Jülich, 2012: 16-27. |
| 28 | PERDEW J P, BURKE K, ERNZERHOF M.. Generalized gradient approximation made simple. Physical Review Letters, 1996, 77 (18): 3865- 3868. |
| 29 | ANDERSEN O K, EBERT H, KOLLAR J, et al. Electronic structure and physical properties of solids: The uses of the LMTO method[C/OL]. Berlin: Springer, 2000[2024-09-27]. https://link.springer.com/book/10.1007/3-540-46437-9. |
| 30 | ANDERSEN O K.. Linear methods in band theory. Physical Review B, 1975, 12 (8): 3060- 3083. |
| 31 | ANDERSEN O K, JEPSEN O.. Explicit, first-principles tight-binding theory. Physical Review Letters, 1984, 53 (27): 2571- 2574. |
| 32 | MARZARI N, MOSTOFI A A, YATES J R, et al.. Maximally localized Wannier functions: Theory and applications. Reviews of Modern Physics, 2012, 84 (4): 1419- 1475. |
| 33 | KRESSE G, FURTHMÜLLER J.. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 1996, 54 (16): 11169- 11186. |
| 34 | MONKHORST H J, PACK J D.. Special points for Brillouin-zone integrations. Physical Review B, 1976, 13 (12): 5188- 5192. |
| 35 | DUDAREV S L, BOTTON G A, SAVRASOV S Y, et al.. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA + U study. Physical Review B, 1998, 57 (3): 1505- 1509. |
| 36 | ANISIMOV V I, ZAANEN J, ANDERSEN O K.. Band theory and Mott insulators: Hubbard U instead of Stoner I. Physical Review B, 1991, 44 (3): 943- 954. |
| 37 | BAUR W H, KHAN A A.. Rutile-type compounds. IV. SiO2, GeO2 and a comparison with other rutile-type structures. Acta Crystallographica Section B, 1971, 27 (11): 2133- 2139. |
| 38 | WANG V, XU N, LIU J C, et al.. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Computer Physics Communications, 2021, 267, 108033. |
| [1] | Runrun DU, Shan WANG, Xuezhi KE. Structural phase transitions of Th2N2S under high pressure: A first-principles calculation study [J]. Journal of East China Normal University(Natural Science), 2024, 2024(3): 36-44. |
| [2] | Wenjie DING, Wenhui XIE. Research on calculation of two-dimensional transition metal chalcogenides compounds MX2-MX-MX2 (M = V, Cr, Mn, and Fe; X = S, Se, and Te) [J]. Journal of East China Normal University(Natural Science), 2024, 2024(3): 45-53. |
| [3] | Jiaojiao LIU, Hongtao LIANG, Yang YANG. Research on atomistic simulation of the coexistence of multiple interfacial states at heterogeneous solid-liquid interface [J]. Journal of East China Normal University(Natural Science), 2024, 2024(3): 54-63. |
| [4] | Wei ZHAO, Qinghong YUAN. Bandgap tuning of C3N: A first-principles study [J]. Journal of East China Normal University(Natural Science), 2022, 2022(4): 114-119. |
| [5] | Yaqiong ZHANG, Wenhui XIE. First-principles calculations investigations of two-dimensional transition metal phosphide MnTn+1(M = V, Cr; T = P, As, and Sb) slices [J]. Journal of East China Normal University(Natural Science), 2022, 2022(2): 84-92. |
| [6] | Qinghua XI, Yiqiang HUANG, Jiaxiang CHEN, Er NIE, Zhuo SUN. Study on Fe2O3/g-C3N4 photocatalytic degradation of Rhodamine B [J]. Journal of East China Normal University(Natural Science), 2021, 2021(3): 151-160. |
| [7] | LI Peng-fei, WANG Mei-ting, MEI Ye. Comparison of the efficiency of equilibrium and nonequilibrium molecular dynamic simulations of molecular solvation free energies [J]. Journal of East China Normal University(Natural Sc, 2019, 2019(1): 83-92. |
| [8] | HUANG Xian-zhi, PIAO Xian-qing, CAI Ya-guo. Preparation of photocatalytic materials MIL-125(Ti)/BiOI and photocatalytic performance study [J]. Journal of East China Normal University(Natural Sc, 2019, 2019(1): 93-104,114. |
| [9] | SHEN Yu-hao, TANG Zheng, PENG Wei. A negatively charged VSiON center for implementation as qubit [J]. Journal of East China Normal University(Natural Sc, 2017, 2017(2): 97-106. |
| [10] | ZHANG Jian-Feng, DAN Shu-Ping. Properties of the bound magnetopolaron in a triangular quantum well [J]. Journal of East China Normal University(Natural Sc, 2015, 2015(6): 101-107. |
| [11] | GAO Li-peng, LIU Kai, LUO Chun-hua, LI Jian-qi,WANG Yi-ting, PENG Hui. Preparation and properties study of a new type of T1-T2 dual mode MRI contrast agent [J]. Journal of East China Normal University(Natural Sc, 2013, 2013(5): 102-109. |
| [12] | SUN Fang, TANG Zheng. Spin interactions in direct-gap semiconductors [J]. Journal of East China Normal University(Natural Sc, 2012, 2012(5): 31-36. |
| [13] | YUAN Li;YANG Xie-long;ZHAO Zhen-jie. Influence of Joule annealing on GMI effect for Co-based amorphous microwires(Chinese) [J]. Journal of East China Normal University(Natural Sc, 2008, 2008(3): 120-124. |
| [14] | WANG Jianqiao, LIU dong, ZHOU Jun, XI Qinghua, NIE Er, SUN Zhuo. Application of TiO2 nanotube arrays for bipolar photocatalytic fuel cells [J]. Journal of East China Normal University(Natural Science), 2020, 2020(1): 93-102. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||